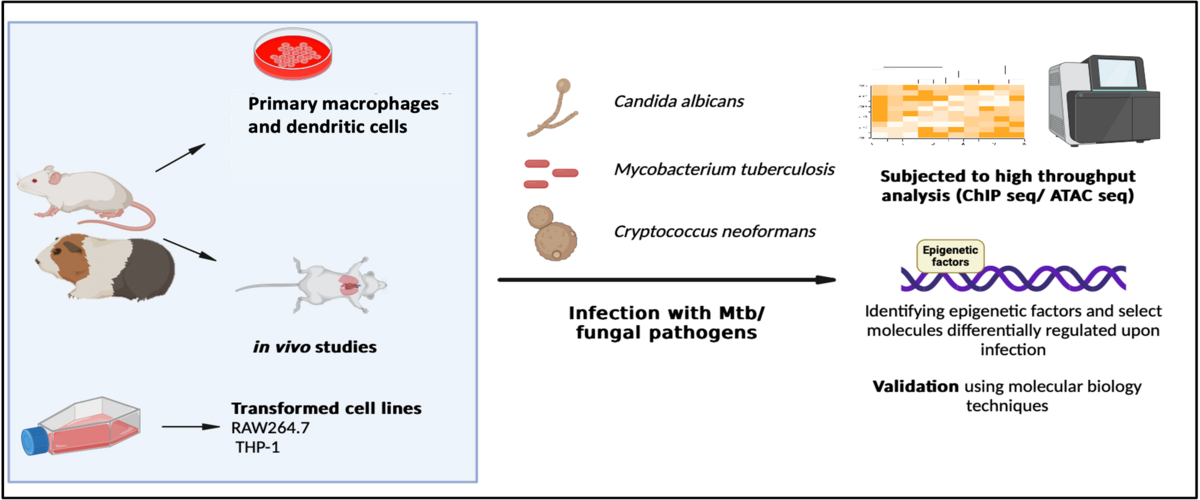

Prof. K. N. Balaji’s lab focuses on the interactions between macrophages and the infectious agents Mycobacterium tuberculosis (Mtb), Candida albicans, and Cryptococcus neoformans. The outcome of an infectious disease is determined by the dynamic interactions between the host and the pathogen. The infectious agent employs distinct immune evasion strategies, modulates the immune signaling of the host, and establishes a niche conducive to its survival. In our laboratory, our primary focus is on delineating the pathogen-driven reprogramming events within sentinel macrophages. We have observed that these events lead to robust changes in the host including lipid accumulation, skewed immune responses; or modulation of host cell pathways such as autophagy, lipophagy, apoptosis, pyroptosis, ferroptosis, etc. Through parallel investigations, we are also studying the pathogen-elicited epigenetic changes within the host and how inhibiting such changes could enhance the efficacy of available therapeutics against the disease.
Mycobacterium tuberculosis, the causative agent of tuberculosis (TB), is an extremely successful intracellular pathogen and a leading killer worldwide. It is estimated to infect nearly one-fourth of the human population and is responsible for 1.3 million deaths annually. Mtb has evolved various immune evasion strategies that can successfully resist and overcome most of the effector functions of its hostile cellular niche. Several mycobacterial effector proteins have been identified to interact with distinct host proteins and undermine the functions of the immune system. Understanding the contribution of each of these mechanisms would be useful in identifying Host-Directed targets against Mtb infection. Our group has previously identified the pivotal role of key immune signaling pathways such as MAPK, JNK, HIPPO, and Sonic Hedgehog in facilitating mycobacterial immune evasion strategies. Recent studies from our group have also unveiled mycobacteria-responsive epigenetic regulation of immune responses identifying key roles for epigenetic factors including JMJD3, BRD4, PRMT5, G9a, and SIRT6 in contributing to mycobacteria-mediated lipid accumulation.
The incidence of fungal infection has surged globally in recent years, with Candida albicans and Cryptococcus neoformans emerging as critical priorities in a recent report by the World Health Organization (WHO). This alarming trend is attributed to the rising population of immunocompromised individuals, driven by shifts in medical practices such as the widespread use of chemotherapy, immunosuppressive drugs, and prosthetic transplants, coupled with the impact of pandemics like HIV and COVID-19. The statistics are sobering, with over 1.5 million deaths and approximately one billion cases of Candida infections reported annually, and around 223,000 cases of Cryptococcal infections resulting in 181,000 deaths. Despite this alarming figure, it has remained a neglected topic by public health authorities. Our research focuses on unraveling the immunological parameters associated with host-fungal interactions across systemic, superficial, and pulmonary infections. By addressing these aspects, we aim to contribute valuable insights to the understanding and potential mitigation of the growing threat posed by fungal infections.
Email : aditisingh@iisc.ac.in
Designation : phd_student
Category : Microbiology, Virology, and Immunology

Email : ashishb@iisc.ac.in
Designation : phd_student
Category : Microbiology, Virology, and Immunology

Email : atheenaa@iisc.ac.in
Designation : phd_student
Category : Microbiology, Virology, and Immunology

Email : awantikashah@iisc.ac.in
Designation : phd_student
Category : Microbiology, Virology, and Immunology

Email : babitakumari@iisc.ac.in
Designation : phd_student
Category : Microbiology, Virology, and Immunology

Email : benasirk@iisc.ac.in
Designation : Support Staff
Category : Microbiology, Virology, and Immunology

Email : sjayashree@iisc.ac.in
Designation : Phd Student
Category : Microbiology, Virology, and Immunology

Email : msamadrita@iisc.ac.in
Designation : Phd Student
Category : Microbiology, Virology, and Immunology

Email : smritisundar@iisc.ac.in
Designation : phd_student
Category : Microbiology, Virology, and Immunology

Email : souravjana@iisc.ac.in
Designation : phd_student
Category : Microbiology, Virology, and Immunology
Cholesterol derived from the host milieu forms a critical factor for mycobacterial pathogenesis. However, the molecular circuitry co-opted by Mycobacterium tuberculosis (Mtb) to accumulate cholesterol in host cells remains obscure. Here, we report that the coordinated action of WNT-responsive histone modifiers G9a (H3K9 methyltransferase) and SIRT6 (H3K9 deacetylase) orchestrate cholesterol build-up in in vitro and in vivo mouse models of Mtb infection. Mechanistically, G9a, along with SREBP2, drives the expression of cholesterol biosynthesis and uptake genes; while SIRT6 along with G9a represses the genes involved in cholesterol efflux. The accumulated cholesterol in Mtb infected macrophages promotes the expression of antioxidant genes leading to reduced oxidative stress, thereby supporting Mtb survival. In corroboration, loss-of-function of G9a in vitro and pharmacological inhibition in vivo; or utilization of BMDMs derived from Sirt6−/− mice or in vivo infection in haplo-insufficient Sirt6−/+mice; hampered host cholesterol accumulation and restricted Mtb burden. These findings shed light on the novel roles of G9a and SIRT6 during Mtb infection and highlight the previously unknown contribution of host cholesterol in potentiating anti-oxidative responses for aiding Mtb survival.
During infection, Mycobacterium tuberculosis (Mtb) rewires distinct host signaling pathways, resulting in pathogen-favorable outcomes. Oxidative stress build-up is a key cellular manifestation that occurs due to the cumulative effect of elevated reactive oxygen species (ROS) generation and the inept ability of the cell to mitigate ROS levels. Here, we report the Mtb-induced expression of the neuronal ligand SLIT2 to be instrumental in ROS accumulation during infection. Loss-of-function analysis revealed the heightened expression of SLIT2 to be dependent on the Mtb-mediated phosphorylation of the P38/JNK pathways. Activation of these kinases resulted in the loss of the repressive H3K27me3 signature on the Slit2 promoter. Furthermore, SLIT2 promoted the expression of Vanin1 (VNN1), which contributed to copious levels of ROS within the host. Thus, we dissect the pathway leading to the robust expression of SLIT2 during Mtb infection while outlining the potential consequences of SLIT2 upregulation in infected macrophages.
Mycobacterium tuberculosis infection leads to the formation of lipid-laden cells (foamy macrophages-FMs) that offer a favorable shelter for its persistence. During infection, we observe a significant reduction in the expression of the E3 ubiquitin ligase, ITCH. This repression allows the sustenance of key lipid accretion molecules (ADRP and CD36), by curbing their proteasomal degradation. Further, we show the repression of ITCH to be dependent on the concerted action of the bifunctional transcription factor, YY1 and the arginine methyl transferase, PRMT5. NOTCH signaling pathway was identified as a master-regulator of YY1 expression. In vitro and in vivo analyses revealed the significance of PRMT5 in regulating FM formation and consequently mycobacterial burden.
Active tuberculosis patients are at high risk of coinfection with opportunistic fungal pathogen Candida albicans. However, the molecular mechanisms that orchestrate pathogenesis of Mycobacterium tuberculosis (Mtb)-C. albicans coinfection remain elusive. In the current study, we utilize a mouse model to demonstrate that Mtb promotes a macrophage environment that is conducive for C. albicans survival. Mtb-dependent protein kinase Cζ-WNT signalling axis induces expression of an E3 ubiquitin ligase, constitutive photomorphogenesis protein 1 (COP1). A secondary infection of C. albicans in such Mtb-infected macrophages causes COP1 to mediate the proteasomal degradation of interferon regulatory factor 9 (IRF9), a cardinal factor that we identified to arbitrate an inflammatory programmed cell death, pyroptosis. In vivo experiments mimicking a pre-existing Mtb infection demonstrate that inhibition of pyroptosis in mice results in increased C. albicans burden and aberrant lung tissue architecture, leading to increased host mortality. Together, our study reveals the crucial role of pyroptosis regulation for manifesting a successful C. albicans–Mtb coinfection.
Mycobacterium tuberculosis (Mtb)-driven lipid accumulation is intricately associated with the progression of tuberculosis (TB) disease. Although several studies elucidating the mechanisms for lipid droplet (LD) biosynthesis exist, we provide evidence for the significance of their regulated turnover via macroautophagy/autophagy during Mtb infection. We demonstrate that Mtb utilizes EGFR (epidermal growth factor receptor) signaling to induce the expression of the histone acetylation reader, BRD4 (bromodomain containing 4). The EGFR-BRD4 axis suppresses lipid-specific autophagy, and hence favors cellular lipid accumulation. Specifically, we found that pharmacological inhibition or knockdown of Egfr or Brd4 enhances autophagic flux and concomitantly decreases cellular LDs that is otherwise maintained at a significant level in chloroquine-treated or Atg5 knocked down autophagy-compromised host cells. In line with the enhanced lipophagy, we found that loss of EGFR or BRD4 function restricts mycobacterial burden that is rescued by external replenishment with oleic acid. We also report that the EGFR-BRD4 axis exerts additional effects by modulating pro-angiogenic gene expression and consequently aberrant angiogenesis during mycobacterial infection. This is important in the context of systemic Mtb dissemination as well as for the efficient delivery of anti-mycobacterial therapeutics to the Mtb-rich core of TB granuloma. Finally, utilizing an in vivo mouse model of TB, we show that pharmacological inhibition of EGFR and BRD4 compromises LD buildup via enhanced lipophagy and normalizes angiogenesis, thereby restricting Mtb burden and rescuing mice from severe TB-like pathology. These findings shed light on the novel roles of BRD4 during Mtb infection, and its possible implication in potentiating anti-TB responses.
Septic arthritis is a chronic inflammatory disorder caused by Staphylococcus aureus invasion of host synovium, which often progresses to impairment of joint functions. Although it is known that disease progression is intricately dependent on dysregulated inflammation of the knee joint, identification of molecular events mediating such imbalance during S. aureus–induced septic arthritis still requires detailed investigation. In this article, we report that Aurora kinase A (AURKA) responsive WNT signaling activates S. aureus infection–triggered septic arthritis, which results in inflammation of the synovium. In this context, treatment with adapalene, a synthetic retinoid derivative, in a mouse model for septic arthritis shows significant reduction of proinflammatory mediators with a simultaneous decrease in bacterial burden and prevents cartilage loss. Mechanistically, adapalene treatment inhibits WNT signaling with concomitant activation of HIPPO signaling, generating alternatively activated macrophages. Collectively, we establish adapalene as a promising strategy to suppress S. aureus–induced irreversible joint damage

gauravohia@alum.iisc.ac.in

salikborbora@alum.iisc.ac.in
preetiyadav@alum.iisc.ac.in
bharatbhatt@alum.iisc.ac.in
tanushreem@alum.iisc.ac.in

ankitag@iisc.ac.in
kasturim@alum.iisc.ac.in
monoranjanb@alum.iisc.ac.in
praveenp@alum.iisc.ac.in
svikas@alum.iisc.ac.in
sahanah@alum.iisc.ac.in
office.mcb@iisc.ac.in
office.mcb@iisc.ac.in
kenhens@hmail.com
office.mcb@iisc.ac.in
bansalk@jncasr.ac.in
nisha2208@gmail.com
a.yash.sinha@gmail.com
dev@nii.ac.in
trinath@hyderabad.bits-pilani.ac.in

mahima16331@iisc.ac.in